

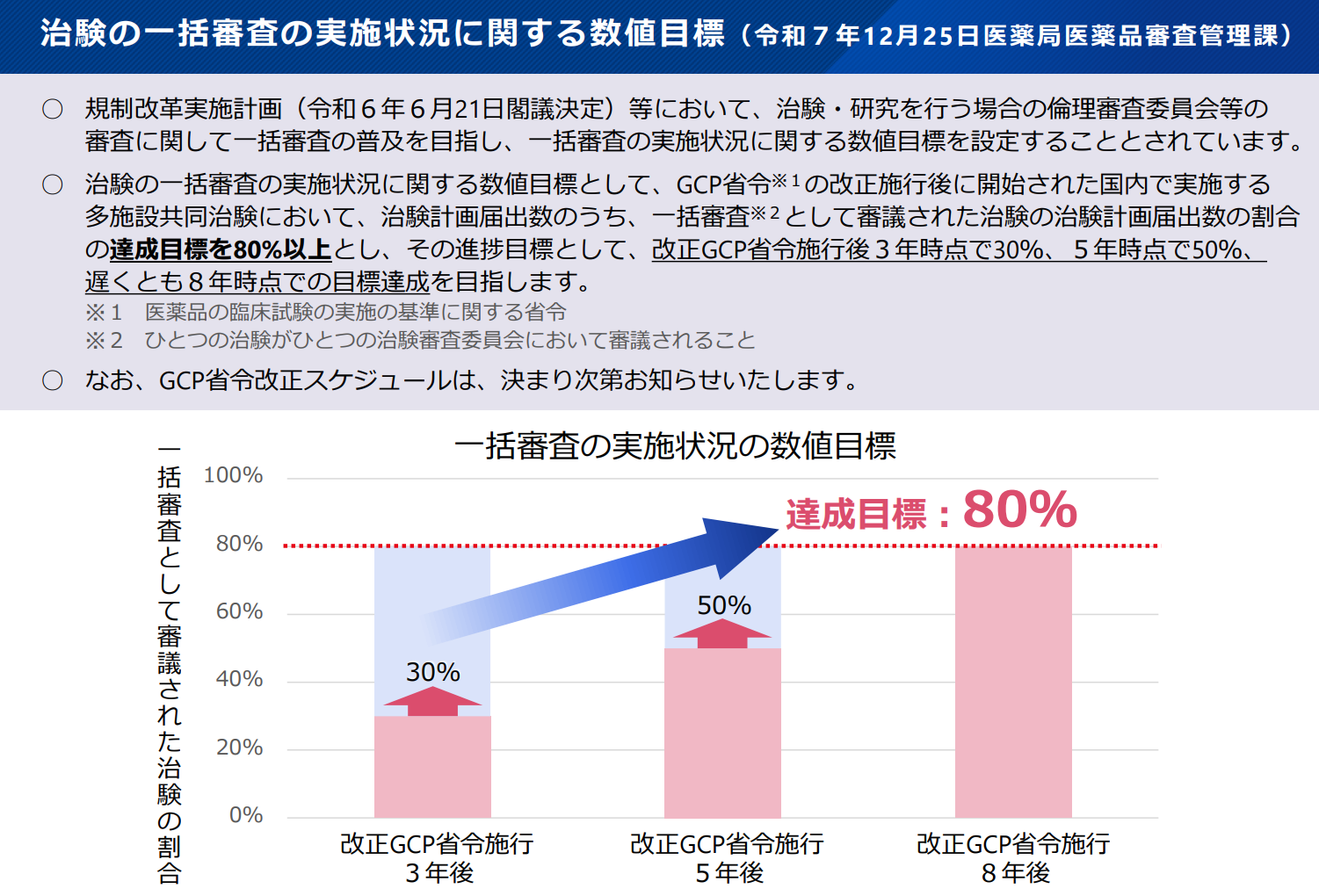
IRBを治験依頼者が選ぶことになる
現在の日本国内ではSingle IRB(1試験で1つの単一の倫理審査委員会)を運用を検討する場合について考えたいと思います。現行のGCP規制(主に厚生労働省令「医薬品の臨床試験の実施の基準(GCP)」、関連通知・Q&A、倫理指針類との整合)を確認すると、日本の現行制度は米国の“sIRB義務化”に相当する包括的な枠組みは未整備で、施設ごとのIRB(1施設で1つのIRB)慣行や、IRB審査の委託・中央化の運用が混在しています。そのため、2027年を見据えると、IRBとの契約・手順面の設計が重要になってきます。
現行制度(2026年改正前)における主な問題点(対応が必要なこと)は以下の通りです。
- 責任主体を明確にする必要: 現行GCPは、スポンサーと治験責任医師/実施医療機関の責務を明確に定め、各医療機関での倫理審査・試験実施体制の適切性を前提にしています。Single IRBを用いる場合でも、各実施医療機関の長(管理者)が当該IRBを治験の受け入れの承認機関として正式に認める体制確認が必要で、実施医療機関側の責任が免除されるわけではありません。
- 外部IRBの利用のSOPの整備が必要: GCPはIRBの構成・独立性・利益相反管理等の要件を規定しています。外部IRB/中央IRBを用いること自体は実務上可能ですが、各施設が「自施設IRBに代えて当該IRBの審査を受ける」ことの院内規程・手順整備が必要で、施設規程によっては外部IRB利用に制限があります。
- 実施医療機関の長(管理者)の最終責任を網羅することが必要: 倫理指針(人を対象とする生命科学・医学系研究倫理指針 等)を参照する観察研究とのハイブリッド運用、つまり企業治験以外の臨床研究では、施設長承認やローカルレビューが実質的に求められる場合があり、Single IRBの形を採っても「施設長決裁」「部門長の安全管理体制確認」など実施医療機関側のローカル承認が残ることがあります。
- 社会的に弱い対象・遺伝学的検査等の特別要件を考量する必要: 小児、妊婦、認知機能低下、遺伝子関連検査・情報取扱いなど、追加的倫理的配慮が必要な試験は、施設固有の臨床倫理委員会関与や専門委員の追加確認が要求される場合があり、Single IRBだけでは完結しにくい場合があります。
今後改正されるGCPはこれらの問題点を解消して、1試験で1つの単一の倫理審査委員会が実施できるような法規制になることが考えられている。そのスキームは「2025年10月15日:あなたが変える治験環境 -ICH-GCP・省令改正と治験エコシステムで描く未来-」で以下の様に示されています。
(引用元:000277591.pdf)
ここでこれからの課題になってくることは、各医療機関での倫理審査・試験実施体制の適切性を前提にしていたことが、治験依頼者(スポンサー)がIRBに直接審査依頼することで、治験依頼者(スポンサー)が倫理審査・試験実施体制の適切性をする必要があることです。
そのため、IRBに対して審査依頼するにあたって、いかに客観的な判断をしてもらうか、その適切をスポンサーがどのように担保していくか、がポイントになってきます。
2025年時点で考えられている方法
IRBの適切性について、前述の2025年10月15日の説明会では以下の様に述べられています。

(引用元:000277591.pdf)
これらのことから、治験依頼者(スポンサー)が適切にSingle IRB選定プロセスを整備していく必要があります。現在考えられている選定プロセスについては、以下の様になっています。
- IRB情報確認シートで各IRBが情報(要件確認シート)を公表する。
- 要件確認シートには、IRB名称、外部機関からの認証情報、開催頻度、委員名簿、利益相反確認方法、治験参加者相談窓口の設置有無、専門性、ゲノム審査の可否などが含まれている。
- 治験依頼者(スポンサー)は要件確認シートと自社の手順を用いて、治験ごとにIRBを選定する。
- IRBと治験依頼者はMSA(Master Service Agreement)を結んで、選定した治験毎に覚書を締結する(予定)。
- 選定して、覚書を締結したIRBを公開(治験計画届に記載やjRCTで公開など)する(予定)。
GCP法改正によって、これたのビュレットのアクションを実行しようとすると、今の社内の体制だと中々難しいのではないでしょうか。2026年はこれから検討していくことが各依頼者沢山ありそうです。
2026年の予想
今後治験依頼者も開発業務を受託するCROも以下のような体制整備が必要なのではないでしょうか。
- 組織体制
- Single IRB方針の確立: どの試験タイプ・被験者層・リスクレベルでSingle IRBを採用するか、例外条件、追加審査が必要な領域(小児、遺伝学的検査等)の方針を明確にする。
- 責任の定義: IRB信頼性担保の管理責任者、IRB審査資料の提出調整役、ICF版管理責任者、安全性配布責任者などの役割を定義する(一部の依頼者やCROによっては既に専門部署を構築済み)。
- IRB選定基準: IRBの適格性要件(委員構成、独立性、専門性、COI、電子審査可否、英文対応など)と評価手順を確立。
- 契約・合意(IRBの信頼性担保と治験実施施設の受入)
- IRB評価の合意(Reliance Agreement): 審査範囲、責任分担、報告線、SLA、記録保存、監査権限、費用、COI管理、電子署名の法的効力を含むテンプレートを整備。
- 治験実施施設側の側受入合意: 各実施医療機関の管理者が外部IRB審査を受容するための覚書/契約条項を準備。院内規程要件(外部IRB利用、個人情報、事故報告)の確認を含む。
- SOP/手順書・品質システムの整備
- Single IRB運用SOP: 提出、照会対応、改訂、継続審査、緊急安全性措置の事後審査、治験主張手続きまでの全プロセスをカバーする。
- 例外・追加審査のハンドリング: 小児・遺伝子・放射線等で必要な施設内追加承認の判定とタイムライン統合。
- 審査運用(提出物、ローカル差分、継続審査)
- 提出物の標準化: プロトコル、IB、ICFマスター+施設版ICF、広告、患者向け資材、補償/治療費説明、のフォーマット化。
- ICFの二層構造: ICFマスターと施設特有条件(謝礼・交通費、相談窓口、費用負担、個人情報問合せ)に分け、相互参照で矛盾防止。
- 軽微改訂・迅速審査: 変更分類基準、迅速審査適用条件の合意。
- 継続審査/年次報告: 審査周期、被験者登録・有害事象の集計様式、継続可否判断の指標を統一。
- 安全性・逸脱・重大事項の報告運用
- 安全性配布計画: SUSAR、DSUR、RSI改訂、IB改訂の配布経路と期限。IRB向けと治験実施施設の院内向けの二層報告を整合させる。
- 重大逸脱の定義と閾値: IRB通知が必要な事象の定義(同意手順不備、無承認実施等)と報告タイムラインを決める。
- 緊急安全性措置: 事前実施→事後IRB報告の手順を標準化し、治験実施施設の院内向け報告の要否もフローを決めておく。
- 文書管理・版管理・トレーサビリティ
- 版管理基盤: すべての審査文書の版・発効日・適用サイトのトラッキング。ICFのマスター/施設版ICFのログ管理でカバーする。
- 承認証跡: IRB議事要旨、決議、条件付き承認事項の解消記録、提出/照会/回答のタイムスタンプ管理。
- 監査・査察・KPI/モニタリング
- IRB適格性監査: 委員教育、COI、SOP、開催記録の確認。必要に応じて現地/リモート監査。
- 査察準備: Single IRBの合理性、例外対応の判断根拠を即時提示できる資料を準備する。
- 費用・請求・予算設計
- 費用スキーム: IRB審査料(初回、改訂、継続)、サイトごとの追加手数料、緊急審査料を見積もり、MSAの契約条件に反映する。
- 経過措置・移行計画(既存試験の取り扱い)
- 移管判断基準: 進行中試験をSingle IRBへ移すか、現行のまま完遂するかの基準(被験者数、残存期間、改訂頻度、サイト数)を決めて、実行する。
- 移行手順: 既存ICF・資材の再承認、同意再取得の要否判断、サイト同意の再取得、タイミングの調整を決める。
特に9の経過措置は業界全体の指針にアラインする必要がありますが、改正GCPの移行目標が以下の様に示されているので、それほど慌てて決めなくても良さそうです。
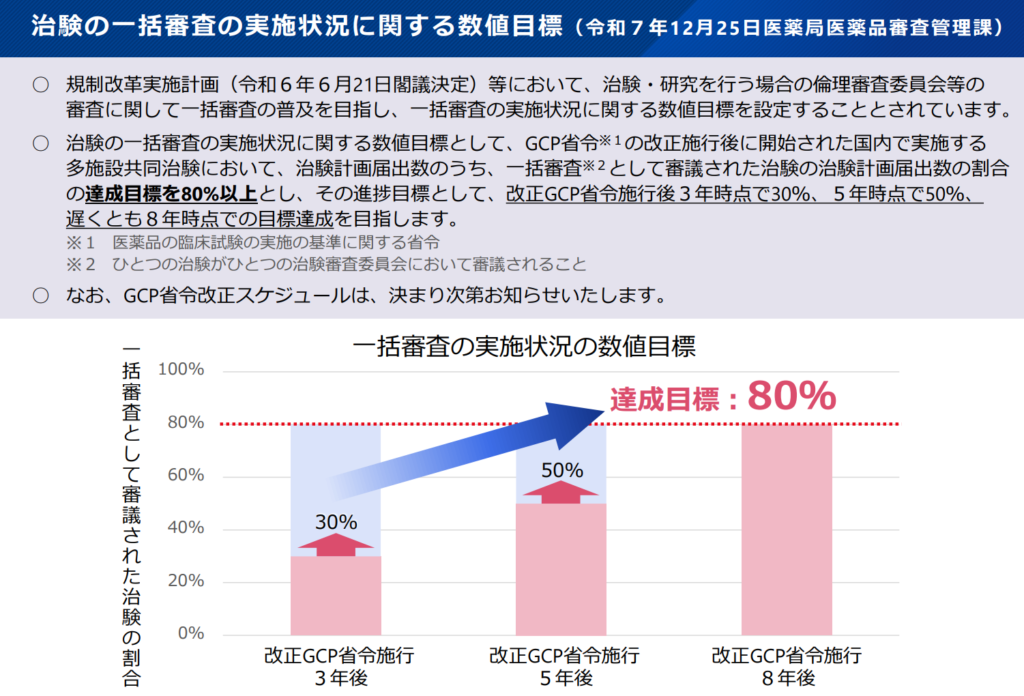
(引用:001622085.pdf)
このように、2026年も治験エコシステム推進のために業界全体で個社も含めて取り組むことが沢山ありそうです。

コメント